Methods
We model things
Mesoscopic scale – Dislocation Dynamics
Thanks to a continuum approach it’s possible to predict the evolution of extended defects in strained semiconductor films. The continuum modeling of dislocation enable one to describe their evolution in films of thickness up to few tenths of microns. We integrate the “MicroMegas” Dislocation Dynamics algorithm with the open-source code for the Finite Elements Method “”, which allows us to describe dislocation evolution in a large variety of conditions, such as multi-layer films, graded semiconductors an so on. Typical process that we are focused on are the evolution of threading dislocations, their interaction and eventually their annihilation.

Atomistic scale – Molecular Dynamics
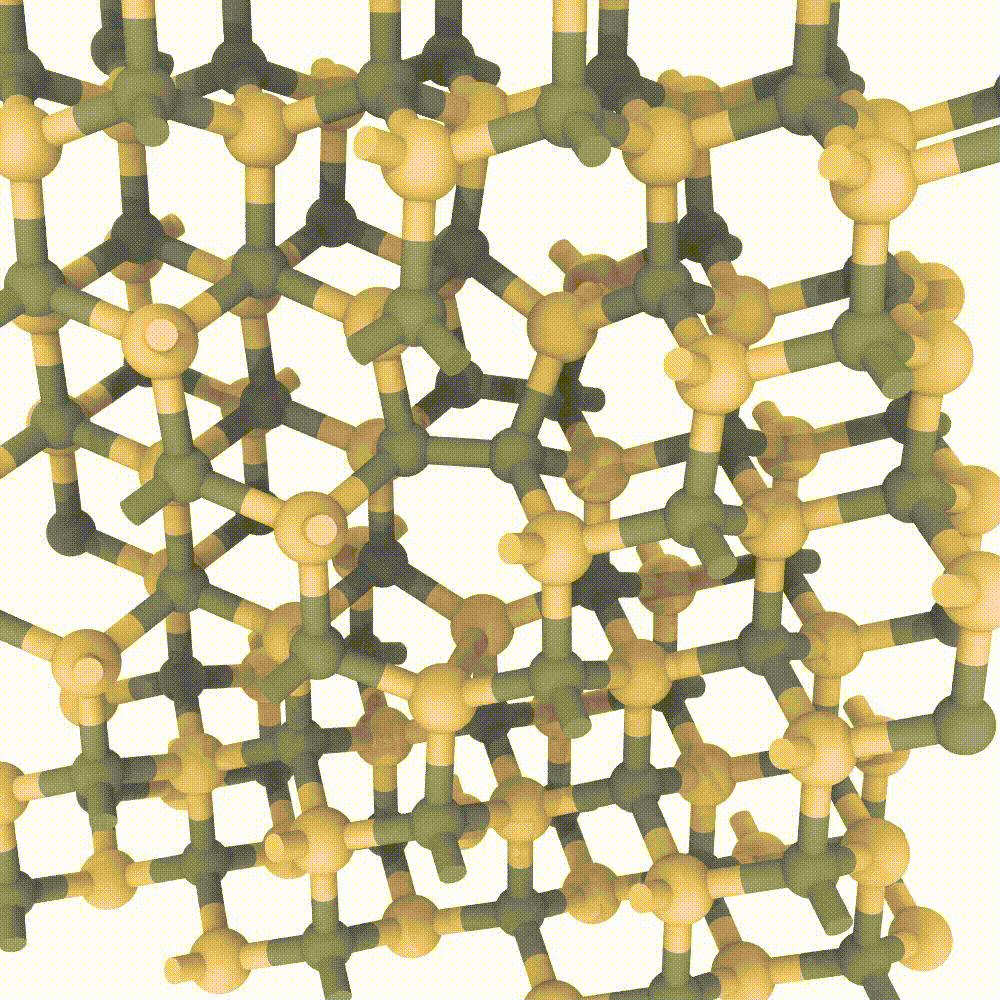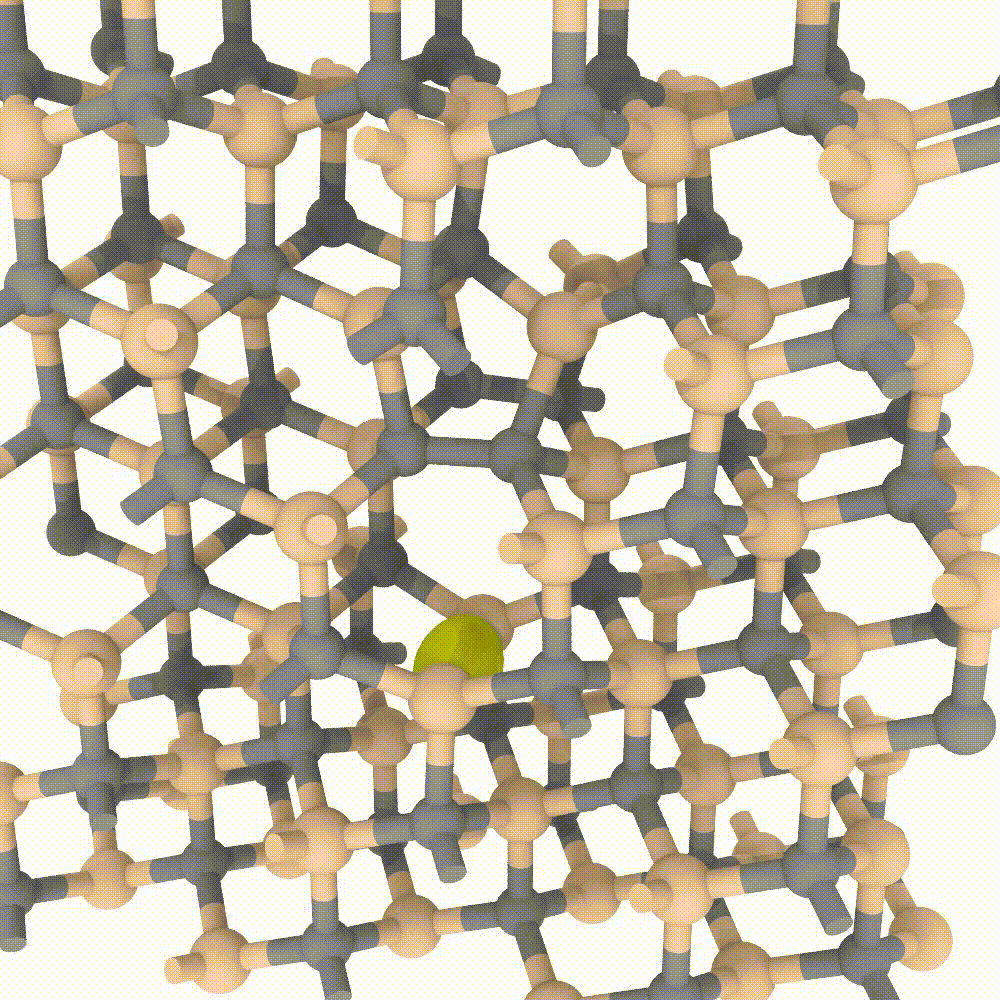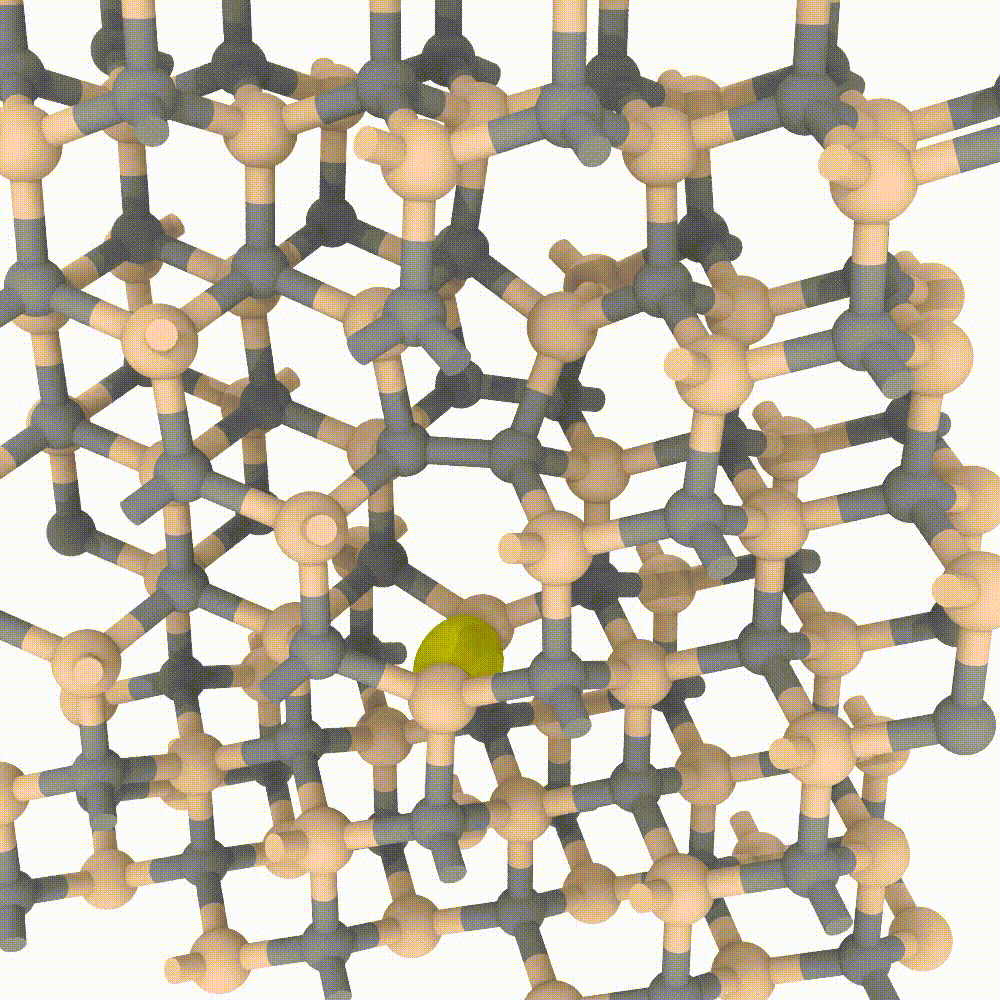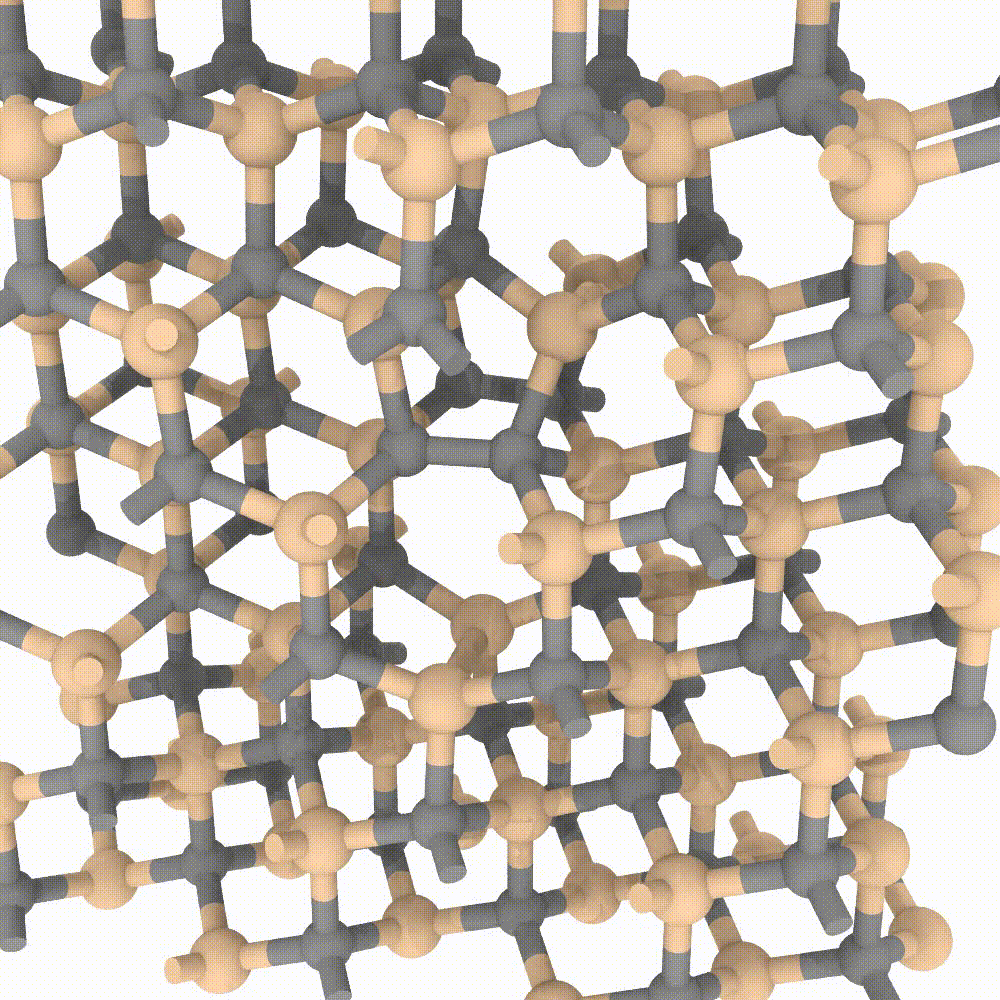
Atomic approaches, aided by the growing speed and power of modern computers, are nowadays able to simulate an incredible amount of particles. In MoSeGroup we employ a classical approach to predict evolution of crystalline defects in supercells composed by an enormous number of atoms (up to 1’000’000 atoms). Many modern codes exist for high performance computing, between them our expertise is focused on the LAMMPS code. A variety of instruments exists that perfectly integrate with LAMMPS simulator; MoSe group employ many of them for the analysis, the visualization and even steering of the simulations.
Electronic properties – Density Functional Theory
The most accurate description of defects involves the understanding of their electronic properties.We exploit the Density Functional Theory to investigate novel materials for nano-electronic and nano-photonic applications. Our activities include the modelling and simulation of semiconductor surfaces, interfaces, and nanostructures, focusing on their structural, vibrational and electronic properties. We employ “Quantum Espresso” and “Vasp” codes for calculation in the framework of Density Functional Theory.
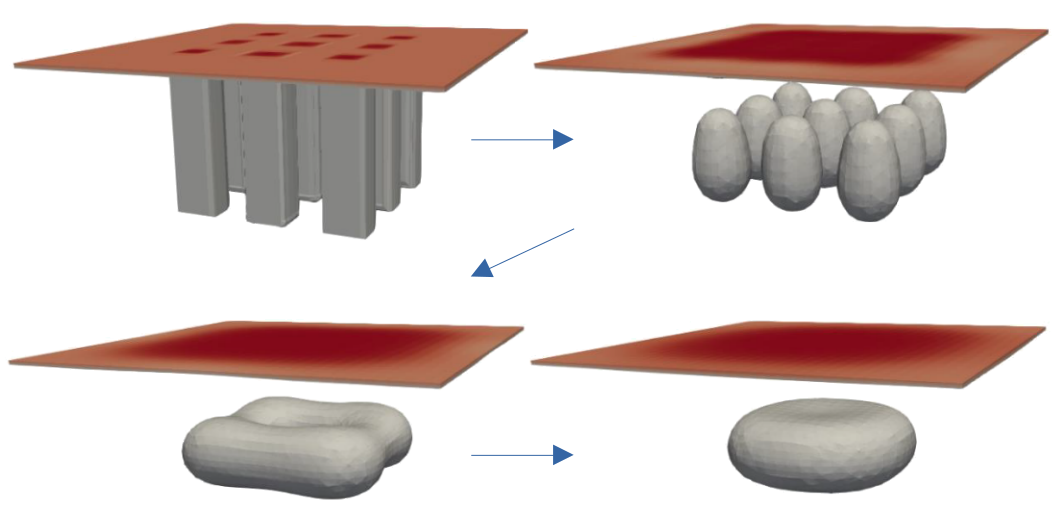
Continuum modelling of crystal growth & phase field
Studying materials on realistic large-size and long-times requires mesoscale modeling which is better suited for a continuum description. In the MoSE group we develop models for the surface evolution dynamics and crystal growth of micro- and nano-structures comprising both thermodynamics and kinetics. The objective is to unveil the complex interplay between crystal faceting, substrate wetting interactions, elastic and plastic deformations, alloy intermixing while materials undergo thermal processing or growth and provide reliable simulations for interpreting the physical behavior and supporting experimental activities. A great part of this research activity exploits the Phase-Field method as a convenient and powerful strategy to tackle arbitrary complex 3D geometries. On a practical ground, continuum simulations generally require the numerical integration of partial differential equations which is performed by different approaches, from finite-difference or spectral methods implemented in home-made codes to finite-element methods eventually based on AMDiS, FEniCS or the commercial software COMSOL Multiphyics.
Machine Learning
Machine Learning is one of the hottest topics in computer science. This surge in popularity also attracted interest in applying these statistical learning approaches to computational physics and materials science. In the MoSe group, we are currently developing novel Machine Learning methods for applications to both continuum and atomistic modelling, in order to push forward the size and time scale tractable by computational methods. At the moment, two main branches of ML algorithms are being studied: using Neural Networks to approximate interatomic potentials coming from DFT calculations in order to speed up molecular dynamics and Recurrent Convolutional Neural Network to approximate non-equilibrium dynamics of mesoscale systems described by continuum approaches.
